Disorders of bile acid synthesis and biliary transport (WP5176)
Homo sapiens

{kind=link}
{kind=link}
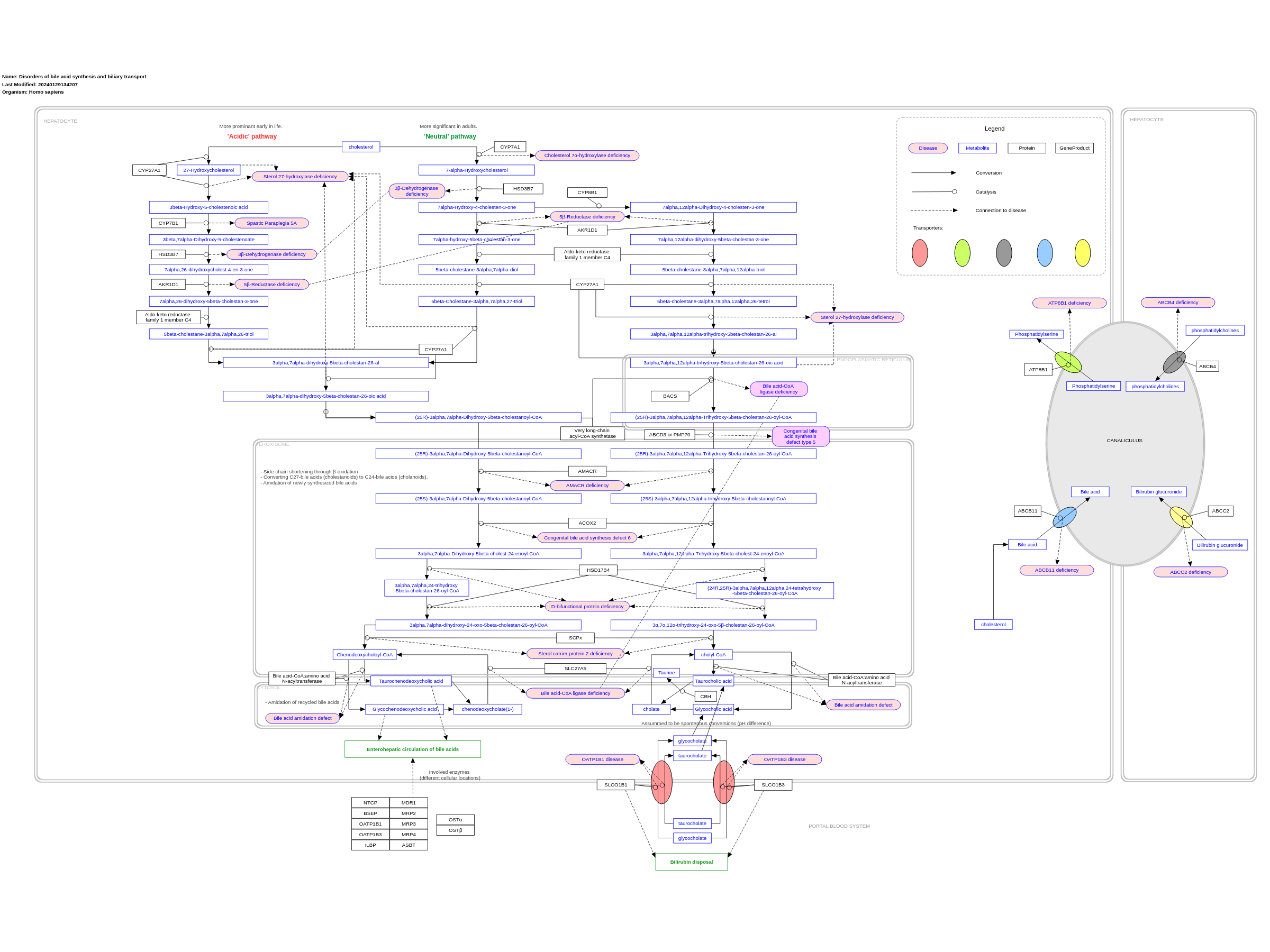
This pathway model displays disorders of bile acid synthesis and biliary transport. Bile acids have a crucial role in the absorption of lipids and hydrophilic vitamins. Furthermore, bile acids aid in the maintenance of cholesterol homeostasis, excretion of toxic substances, processing of food intake, and used as signaling molecules influencing glucose homeostasis, lipid metabolism, and energy expenditure. First, bile acids with a low solubility (less hydrophilic, unconjugated) must be activated using CoA, so that conjugation to taurine or glycine can happen. This model includes 20 disorders, of which 14 are enzyme deficiencies, and 6 are related to transporters. The enzyme deficiencies include 3β-Dehydrogenase deficiency, 5β-Reductase deficiency, Spastic Paraplegia 5A, Cholesterol 7α-hydroxylase deficiency, Sterol 27-hydroxylase deficiency, α-Methylacyl-CoA racemase (AMACR) deficiency, Bile acid amidation defect, and Bile acid-CoA ligase deficiency (BA CoA LD, BACS), congenital bile acid synthesis defect 6 and type 5, D-bifunctional protein deficiency, sterol carrier protein 2 deficiency. Disorders of transporters are related to deficiencies in ATP8B1 (Progressive familial intrahepatic cholestasis type 1; PFIC1), ABCB11 (Progressive familial intrahepatic cholestasis type 2; PFIC 2), ABCB4 (Progressive familial intrahepatic cholestasis type 3), and ABCC2 (Dubin-Johnson syndrome), as well as Rotor Syndrome (linked to two distinct genes, OATP1B1 and OATP1B3, related to taurocholate and glycocholate transport). This pathway is based on Chapter 34 of Blau’s ‘Physicians Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases (ISBN 3642403360 (978-3642403361)), edition 4 (and is currently in the process of being updated to edition 5, Chapter 56). We would like to thank two authors from this chapter (Frédéric M. Vaz and Sacha Ferdinandusse) for their efforts in curating this pathway model!
For a description of pathway objects, see the WikiPathways Legend.
Authors
Maria van de Meent , Eric Weitz , Denise Slenter , and Egon WillighagenActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways ONTOX Rare Diseases Serious Request 2024 - MetaKidsAnnotations
Disease Ontology
progressive familial intrahepatic cholestasis 3 progressive familial intrahepatic cholestasis 2 progressive familial intrahepatic cholestasis 1 cerebrotendinous xanthomatosis congenital bile acid synthesis defect hereditary spastic paraplegia 5A alpha-methylacyl-CoA racemase deficiency Dubin-Johnson syndromePathway Ontology
disease pathway inborn error of metabolism pathway bile acid transport pathway transport pathway bile acid biosynthetic pathway primary bile acid biosynthetic pathway secondary bile acid biosynthetic pathwayCell Type Ontology
hepatocyte| Label | Type | Compact URI | Comment |
|---|---|---|---|
| 3alpha,7alpha,12alpha-Trihydroxy-5beta-cholest-24-enoyl-CoA | Metabolite | chebi:27505 | |
| (24R,25R)-3alpha,7alpha,12alpha,24-tetrahydroxy-5beta-cholestan-26-oyl-CoA | Metabolite | chebi:52050 | |
| 3alpha,7alpha-dihydroxy-5beta-cholestan-26-oic acid | Metabolite | chebi:16577 | |
| 3alpha,7alpha,24-trihydroxy-5beta-cholestan-26-oyl-CoA | Metabolite | wikidata:Q27103111 | |
| 3alpha,7alpha-Dihydroxy-5beta-cholest-24-enoyl-CoA | Metabolite | chebi:27393 | |
| glycocholate | Metabolite | chebi:29746 | |
| taurocholate | Metabolite | chebi:36257 | AKA cholyltaurine |
| 3alpha,7alpha,12alpha-trihydroxy-5beta-cholestan-26-oic acid | Metabolite | chebi:18402 | AKA THCA, trihydroxycholestanoic acid, 3α,7α,12α-trihydroxy-5β-cholestanoic acid |
| 3alpha,7alpha-dihydroxy-24-oxo-5beta-cholestan-26-oyl-CoA | Metabolite | chebi:87704 | |
| Taurine | Metabolite | chebi:507393 | |
| 3α,7α,12α-trihydroxy-24-oxo-5β-cholestan-26-oyl-CoA | Metabolite | chebi:27379 | |
| (25S)-3alpha,7alpha,12alpha-trihydroxy-5beta-cholestanoyl-CoA | Metabolite | chebi:37643 | |
| 5beta-cholestane-3alpha,7alpha,26-triol | Metabolite | chebi:28540 | |
| 3alpha,7alpha,12alpha-trihydroxy-5beta-cholestan-26-al | Metabolite | chebi:16466 | |
| 3alpha,7alpha-dihydroxy-5beta-cholestan-26-al | Metabolite | chebi:27428 | |
| 5beta-cholestane-3alpha,7alpha,12alpha,26-tetrol | Metabolite | chebi:17278 | |
| 7alpha,26-dihydroxycholest-4-en-3-one | Metabolite | chebi:48825 | |
| 7alpha,26-dihydroxy-5beta-cholestan-3-one | Metabolite | chebi:48778 | |
| Phosphatidylserine | Metabolite | chebi:18303 | |
| 3beta-Hydroxy-5-cholestenoic acid | Metabolite | chebi:81014 | |
| Bile acid | Metabolite | chebi:36277 | |
| phosphatidylcholines | Metabolite | chebi:49183 | |
| Bilirubin glucuronide | Metabolite | chebi:16427 | |
| chenodeoxycholate(1-) | Metabolite | chebi:36234 | |
| cholesterol | Metabolite | chebi:16113 | |
| 7-alpha-Hydroxycholesterol | Metabolite | chebi:17500 | |
| 7alpha,12alpha-Dihydroxy-4-cholesten-3-one | Metabolite | chebi:28477 | |
| (25S)-3alpha,7alpha-Dihydroxy-5beta-cholestanoyl-CoA | Metabolite | hmdb:HMDB0060306 | |
| 5beta-cholestane-3alpha,7alpha,12alpha-triol | Metabolite | chebi:16496 | |
| Glycocholic acid | Metabolite | chebi:17687 | |
| (25R)-3alpha,7alpha-Dihydroxy-5beta-cholestanoyl-CoA | Metabolite | hmdb:HMDB0060304 | |
| cholyl-CoA | Metabolite | chebi:15519 | |
| 3beta,7alpha-Dihydroxy-5-cholestenoate | Metabolite | hmdb:HMDB0012454 | Alternative name: 3β,7α-Dihydroxy-5-cholestenoic acid |
| Taurocholic acid | Metabolite | chebi:36257 | |
| Chenodeoxycholoyl-CoA | Metabolite | chebi:28701 | |
| 7alpha,12alpha-dihydroxy-5beta-cholestan-3-one | Metabolite | chebi:2288 | |
| 5beta-cholestane-3alpha,7alpha-diol | Metabolite | chebi:28047 | |
| cholate | Metabolite | chebi:29747 | |
| 27-Hydroxycholesterol | Metabolite | chebi:17703 | C27-carboxylic acid |
| 7alpha-hydroxy-5beta-cholestan-3-one | Metabolite | chebi:2290 | |
| 7alpha-Hydroxy-4-cholesten-3-one | Metabolite | chebi:17899 | |
| Glycochenodeoxycholic acid | Metabolite | chebi:36274 | |
| Taurochenodeoxycholic acid | Metabolite | chebi:16525 | |
| 5beta-Cholestane-3alpha,7alpha,27-triol | Metabolite | hmdb:HMDB0060138 | Alternative name: 5β-Cholestan-3α,7α,27-triol |
| (25R)-3alpha,7alpha,12alpha-Trihydroxy-5beta-cholestan-26-oyl-CoA | Metabolite | chebi:37642 | AKA THC-CoA |
| SLCO1B1 | GeneProduct | uniprot:Q9Y6L6 | |
| SLCO1B3 | GeneProduct | uniprot:Q9NPD5 | |
| ACOX2 | GeneProduct | ensembl:ENSG00000168306 | |
| SCPx | GeneProduct | uniprot:P22307 | Isoforms SCP2 and SCPx cooperate in peroxisomal oxidation of certain naturally occurring tetramethyl-branched fatty acyl-CoA [Source: UniProt] |
| ABCB11 | GeneProduct | ensembl:ENSG00000073734 | |
| ABCC2 | GeneProduct | ensembl:ENSG00000023839 | |
| SLC27A5 | GeneProduct | ensembl:ENSG00000083807 | |
| CYP7A1 | GeneProduct | ensembl:ENSG00000167910 | aka sterol 7α-hydroxylase |
| ABCB4 | GeneProduct | ensembl:ENSG00000005471 | |
| CYP7B1 | GeneProduct | ensembl:ENSG00000172817 | |
| HSD3B7 | GeneProduct | ensembl:ENSG00000099377 | |
| AKR1D1 | GeneProduct | ensembl:ENSG00000122787 | |
| CYP8B1 | GeneProduct | uniprot:Q9UNU6 | |
| AMACR | GeneProduct | uniprot:Q9UHK6 | |
| ATP8B1 | GeneProduct | ensembl:ENSG00000081923 | |
| CYP27A1 | GeneProduct | ensembl:ENSG00000135929 | AKA sterol 27-hydroxylase |
| CYP27A1 | GeneProduct | ensembl:ENSG00000135929 | |
| HSD17B4 | GeneProduct | ensembl:ENSG00000133835 | |
| BACS | Protein | uniprot:Q9Y2P5 | AKA Bile acid-CoA ligase, gene symbol: SLC27A5, Long-chain fatty acid transport protein 5 |
| CBH | Protein | uniprot:P54965 | AKA conjugated bile acid hydrolase, or CBAH, bacterial conversionTentatively annotated for species Clostridium perfringens (strain 13 / Type A), C. perfringens, based on Table 1 from PMID: 30098054 |
| Aldo-keto reductase family 1 member C4 | Protein | uniprot:P17516 | |
| Very long-chain acyl-CoA synthetase | Protein | uniprot:O14975 | Full name: very long chain acyl-CoA synthase (VLCS)/di-/trihydroxycholestanoic acid-CoA ligaseCheck annotation of this enzyme! |
| ABCD3 or PMP70 | Protein | uniprot:P28288 | |
| Bile acid-CoA:amino acid N-acyltransferase | Protein | uniprot:Q14032 | amino acid N-acyltransferase (BAAT), which has a dual localization (peroxisome and cytosol) |
| NTCP | Protein | uniprot:Q14973 | AKA SLC10A1 |
| OATP1B1 | Protein | uniprot:Q9Y6L6 | AKA SLCO1B1 |
| OATP1B3 | Protein | uniprot:Q9NPD5 | AKA SLCO1B3 |
| MRP2 | Protein | uniprot:Q92887 | AKA ABCC2 |
| MRP3 | Protein | uniprot:O15438 | AKA ABCC3 |
| BSEP | Protein | uniprot:O95342 | AKA ABCB11 |
| MRP4 | Protein | uniprot:O15439 | AKA ABBC4 (PMID:21103970); according to UniProt ABCC4 is the correct gene name. |
| OSTα | Protein | uniprot:Q86UW1 | heteromeric transporter OSTα-OSTβ , assumed to be gene SLC51A based on UniProt entry (gene name not mentioned in publication) |
| OSTβ | Protein | uniprot:Q86UW2 | heteromeric transporter OSTα-OSTβ , assumed to be gene SLC51B based on UniProt entry (gene name not mentioned in publication) |
| MDR1 | Protein | uniprot:P08183 | AKA ABCB1A (PMID:21103970), UniProt: ABCB1 (ABCB1A is used for mouse). MDR1A in Figure (PMID:21103970) |
| ILBP | Protein | uniprot:P51161 | AKA FABP6 |
| ASBT | Protein | uniprot:Q12908 | AKA SLC10A2 |
References
- Familial giant cell hepatitis associated with synthesis of 3 beta, 7 alpha-dihydroxy-and 3 beta,7 alpha, 12 alpha-trihydroxy-5-cholenoic acids. Clayton PT, Leonard JV, Lawson AM, Setchell KD, Andersson S, Egestad B, et al. J Clin Invest. 1987 Apr;79(4):1031–8. PubMed Europe PMC Scholia
- Delta 4-3-oxosteroid 5 beta-reductase deficiency and neonatal hemochromatosis. Clayton PT. J Pediatr. 1994 Nov;125(5 Pt 1):845–6. PubMed Europe PMC Scholia
- Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. Setchell KD, Schwarz M, O’Connell NC, Lund EG, Davis DL, Lathe R, et al. J Clin Invest. 1998 Nov 1;102(9):1690–703. PubMed Europe PMC Scholia
- Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Ferdinandusse S, Denis S, Clayton PT, Graham A, Rees JE, Allen JT, et al. Nat Genet. 2000 Feb;24(2):188–91. PubMed Europe PMC Scholia
- Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Verrips A, Hoefsloot LH, Steenbergen GC, Theelen JP, Wevers RA, Gabreëls FJ, et al. Brain. 2000 May;123 ( Pt 5):908–19. PubMed Europe PMC Scholia
- Participation of two members of the very long-chain acyl-CoA synthetase family in bile acid synthesis and recycling. Mihalik SJ, Steinberg SJ, Pei Z, Park J, Kim DG, Heinzer AK, et al. J Biol Chem. 2002 Jul 5;277(27):24771–9. PubMed Europe PMC Scholia
- Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, et al. J Clin Invest. 2002 Jul;110(1):109–17. PubMed Europe PMC Scholia
- Liver disease caused by failure to racemize trihydroxycholestanoic acid: gene mutation and effect of bile acid therapy. Setchell KDR, Heubi JE, Bove KE, O’Connell NC, Brewsaugh T, Steinberg SJ, et al. Gastroenterology. 2003 Jan;124(1):217–32. PubMed Europe PMC Scholia
- Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. Clayton PT, Verrips A, Sistermans E, Mann A, Mieli-Vergani G, Wevers R. J Inherit Metab Dis. 2002 Oct;25(6):501–13. PubMed Europe PMC Scholia
- A novel HPLC-based method to diagnose peroxisomal D-bifunctional protein enoyl-CoA hydratase deficiency. Gloerich J, Denis S, van Grunsven EG, Dacremont G, Wanders RJA, Ferdinandusse S. J Lipid Res. 2003 Mar;44(3):640–4. PubMed Europe PMC Scholia
- Mutations in SRD5B1 (AKR1D1), the gene encoding delta(4)-3-oxosteroid 5beta-reductase, in hepatitis and liver failure in infancy. Lemonde HA, Custard EJ, Bouquet J, Duran M, Overmars H, Scambler PJ, et al. Gut. 2003 Oct;52(10):1494–9. PubMed Europe PMC Scholia
- Diet, anaerobic bacterial metabolism, and colon cancer: a review of the literature. McGarr SE, Ridlon JM, Hylemon PB. J Clin Gastroenterol. 2005 Feb;39(2):98–109. PubMed Europe PMC Scholia
- Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Ferdinandusse S, Kostopoulos P, Denis S, Rusch H, Overmars H, Dillmann U, et al. Am J Hum Genet. 2006 Jun;78(6):1046–52. PubMed Europe PMC Scholia
- Relapsing encephalopathy in a patient with alpha-methylacyl-CoA racemase deficiency. Thompson SA, Calvin J, Hogg S, Ferdinandusse S, Wanders RJA, Barker RA. J Neurol Neurosurg Psychiatry. 2008 Apr;79(4):448–50. PubMed Europe PMC Scholia
- Variable clinical spectrum of the most common inborn error of bile acid metabolism--3beta-hydroxy-Delta 5-C27-steroid dehydrogenase deficiency. Subramaniam P, Clayton PT, Portmann BC, Mieli-Vergani G, Hadzić N. J Pediatr Gastroenterol Nutr. 2010 Jan;50(1):61–6. PubMed Europe PMC Scholia
- Differences in presentation and progression between severe FIC1 and BSEP deficiencies. Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, et al. J Hepatol. 2010 Jul;53(1):170–8. PubMed Europe PMC Scholia
- Typical cMRI pattern as diagnostic clue for D-bifunctional protein deficiency without apparent biochemical abnormalities in plasma. Grønborg S, Krätzner R, Spiegler J, Ferdinandusse S, Wanders RJA, Waterham HR, et al. Am J Med Genet A. 2010 Nov;152A(11):2845–9. PubMed Europe PMC Scholia
- Clinical phenotype variability in patients with hereditary spastic paraplegia type 5 associated with CYP7B1 mutations. Arnoldi A, Crimella C, Tenderini E, Martinuzzi A, D’Angelo MG, Musumeci O, et al. Clin Genet. 2012 Feb;81(2):150–7. PubMed Europe PMC Scholia
- Disorders of bile acid synthesis. Clayton PT. J Inherit Metab Dis. 2011 Jun;34(3):593–604. PubMed Europe PMC Scholia
- Bile acid-CoA ligase deficiency--a new inborn error of bile acid metabolism. Chong CPK, Mills PB, McClean P, Gissen P, Bruce C, Stahlschmidt J, et al. J Inherit Metab Dis. 2012 May;35(3):521–30. PubMed Europe PMC Scholia
- Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. van de Steeg E, Stránecký V, Hartmannová H, Nosková L, Hřebíček M, Wagenaar E, et al. J Clin Invest. 2012 Feb;122(2):519–28. PubMed Europe PMC Scholia
- Diagnosis in bile acid-CoA: amino acid N-acyltransferase deficiency. Hadžić N, Bull LN, Clayton PT, Knisely AS. World J Gastroenterol. 2012 Jul 7;18(25):3322–6. PubMed Europe PMC Scholia
- Specific combination of compound heterozygous mutations in 17β-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency. McMillan HJ, Worthylake T, Schwartzentruber J, Gottlieb CC, Lawrence SE, Mackenzie A, et al. Orphanet J Rare Dis. 2012 Nov 22;7:90. PubMed Europe PMC Scholia
- ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Vilarinho S, Sari S, Mazzacuva F, Bilgüvar K, Esendagli-Yilmaz G, Jain D, et al. Proc Natl Acad Sci U S A. 2016 Oct 4;113(40):11289–93. PubMed Europe PMC Scholia
- ACOX2 deficiency: An inborn error of bile acid synthesis identified in an adolescent with persistent hypertransaminasemia. Monte MJ, Alonso-Peña M, Briz O, Herraez E, Berasain C, Argemi J, et al. J Hepatol. 2017 Mar;66(3):581–8. PubMed Europe PMC Scholia
- Bile acid analysis in human disorders of bile acid biosynthesis. Vaz FM, Ferdinandusse S. Mol Aspects Med. 2017 Aug;56:10–24. PubMed Europe PMC Scholia
- Bile salt hydrolases: Structure and function, substrate preference, and inhibitor development. Dong Z, Lee BH. Protein Sci. 2018 Oct;27(10):1742–54. PubMed Europe PMC Scholia
- Dubin Johnson Syndrome. Talaga ZJ, Vaidya PN. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. PubMed Europe PMC Scholia
- Epidemiology and burden of progressive familial intrahepatic cholestasis: a systematic review. Jones-Hughes T, Campbell J, Crathorne L. Orphanet J Rare Dis. 2021 Jun 3;16(1):255. PubMed Europe PMC Scholia