Disorders of galactose metabolism (WP5173)
Homo sapiens

{kind=link}
{kind=link}
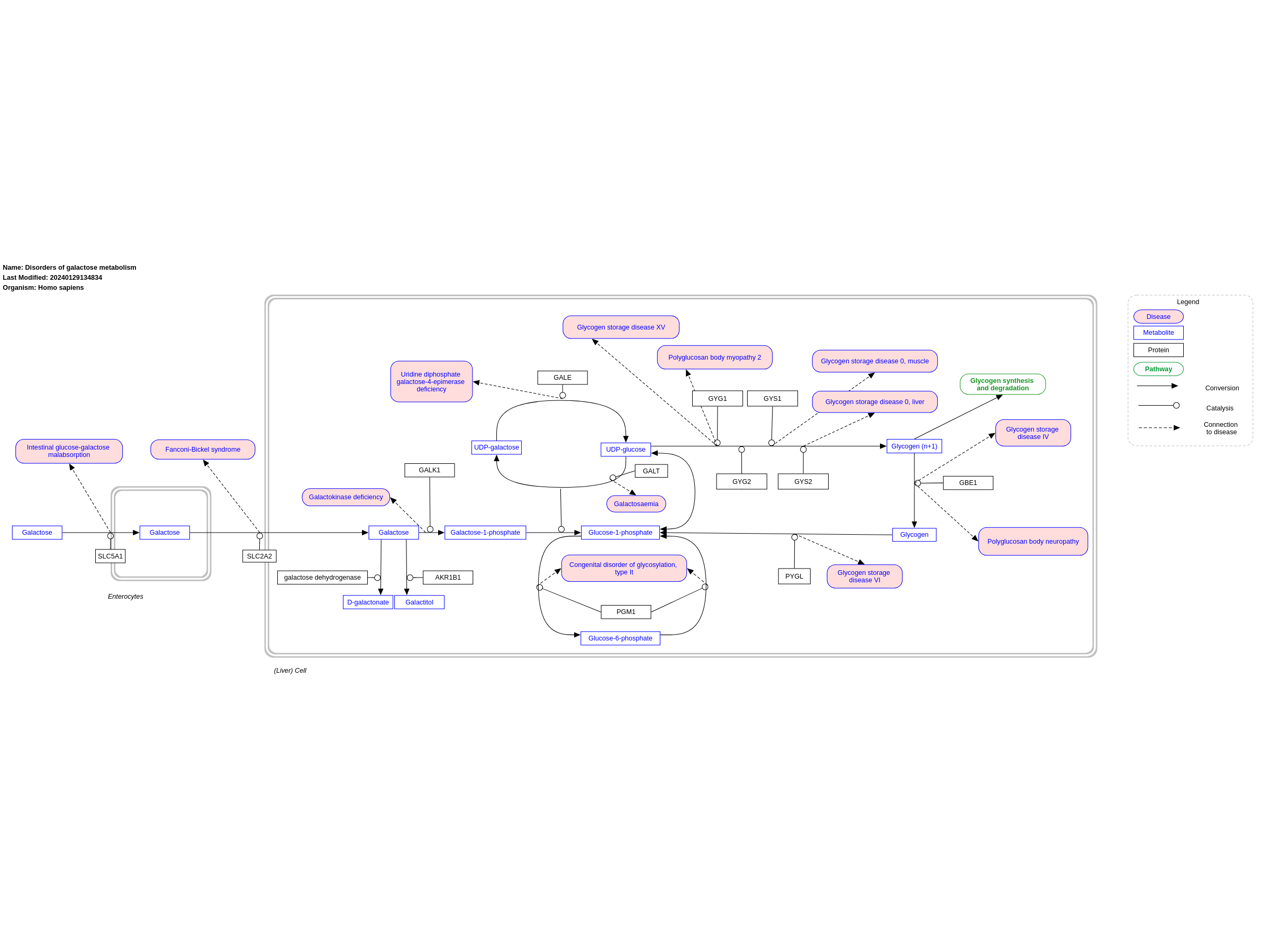
Galactose is converted into glucose 1-phosphate (G1P) through a series of steps called the Leloir pathway. The first step of the pathway is the phosphorylation of galactose by galactokinase (encoded GALK1) to yield galactose 1-phosphate. Conversion of galactose 1-phosphate to G1P requires the transfer of UDP from UDP-glucose catalyzed by GALT. UDP-galactose is converted to UDP-glucose by GALE. Glucose-1-phosphate is converted to glucose-6-phosphate by phosphoglucomutase (PGM) and vice versa. There are two known disorders concerning the uptake transports of galactose (SGLT1 and GLUT2 deficiency) and three known disorders of galactose metabolism: galactokinase deficiency (GALK-D), galactose 1-phosphate uridyltransferase deficiency (galactosemia, GALT-D) and uridine diphosphate galactose 4-epimerase deficiency (GALE-D). Among these, galactosemia is the most common and most severe. This pathway was inspired by Chapter 18, figure 18.3 of the book of Blau (4th edition; ISBN: 978-3-642-40337-8).
For a description of pathway objects, see the WikiPathways Legend.
Authors
Alexandra Bosch , Enzo Chiaradia , Egon Willighagen , Denise Slenter , Lars Willighagen , and Eric WeitzActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways ONTOX Rare Diseases Serious Request 2024 - MetaKidsAnnotations
Disease Ontology
glycogen storage disease XV galactosemia glycogen storage disease VI galactose epimerase deficiency congenital disorder of glycosylation It glycogen storage disease galactokinase deficiency glycogen storage disease IVPathway Ontology
GALE deficiency pathway carbohydrate metabolic pathway galactose metabolic pathway classic metabolic pathway glycolysis pathway disease pathway altered galactose metabolic pathway| Label | Type | Compact URI | Comment |
|---|---|---|---|
| Glycogen (n+1) | Metabolite | cas:9005-79-2 | |
| Glycogen | Metabolite | chebi:28087 | |
| Glucose-1-phosphate | Metabolite | chebi:16077 | |
| Galactitol | Metabolite | hmdb:HMDB0000107 | |
| UDP-galactose | Metabolite | chebi:67119 | |
| UDP-glucose | Metabolite | chebi:18066 | |
| Galactose | Metabolite | chebi:28260 | |
| Galactose-1-phosphate | Metabolite | chebi:17973 | |
| Glucose-6-phosphate | Metabolite | hmdb:HMDB0001401 | search for glucose 6-phosphate |
| D-galactonate | Metabolite | chebi:12931 | |
| GYS1 | GeneProduct | ensembl:ENSG00000104812 | catalyzes the rate-limiting step in glycogen synthesis in the liver and in skeletal muscle: the transfer of glucose monomers from UDP-glucose to the terminal branch of the growing glycogen chain via the formation of α(1→4) glycosidic bondsGYS1: specific to skeletal muscle |
| GBE1 | GeneProduct | ensembl:ENSG00000114480 | glycogen branching enzyme that catalyzes the transfer of alpha-1,4-linked glucosyl units from the outer end of a glycogen chain to an alpha-1,6 position on the same or a neighboring glycogen chain |
| PYGL | GeneProduct | ensembl:ENSG00000100504 | |
| GYG2 | GeneProduct | ensembl:ENSG00000056998 | expressed mainly in liver, cardiac muscle and other types of tissue, but not in skeletal muscle. |
| GYG1 | GeneProduct | ensembl:ENSG00000163754 | Glycogenin-1 is involved in the biosynthesis of glycogen. It is capable of self-glucosylation, forming an oligosaccharide primer that serves as a substrate for glycogen synthase. It also plays a role in glycogen metabolism regulationexpressed mostly in muscles |
| GYS2 | GeneProduct | ensembl:ENSG00000111713 | GYS2: specific to liver |
| AKR1B1 | GeneProduct | ensembl:ENSG00000085662 | |
| GALE | GeneProduct | ensembl:ENSG00000117308 | |
| PGM1 | GeneProduct | ensembl:ENSG00000079739 | |
| GALT | GeneProduct | ensembl:ENSG00000213930 | |
| SLC5A1 | GeneProduct | ensembl:ENSG00000100170 | |
| GALK1 | GeneProduct | ensembl:ENSG00000108479 | |
| SLC2A2 | GeneProduct | ensembl:ENSG00000163581 | |
| Galactosedehydrogenase | Protein | eccode:1.1.1.48 | 'This enzyme is part of the De Ley-Doudoroff pathway, which is used by some bacteria during growth on D-galactose.' Source: [https://enzyme.expasy.org/EC/1.1.1.48] |
References
- Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases [Internet]. Blau N, Duran M, Gibson KM, Dionisi Vici C, editors. Springer Berlin Heidelberg; 2014. Available from: http://dx.doi.org/10.1007/978-3-642-40337-8 DOI Scholia
- Hypoglycemia. Koren D, Palladino A. In: Genetic Diagnosis of Endocrine Disorders [Internet]. Elsevier; 2016. p. 31–75. Available from: http://dx.doi.org/10.1016/B978-0-12-800892-8.00003-8 DOI Scholia
- Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Morava E. Mol Genet Metab. 2014 Aug;112(4):275–9. PubMed Europe PMC Scholia
- Galactose metabolism and health. Coelho AI, Berry GT, Rubio-Gozalbo ME. Curr Opin Clin Nutr Metab Care. 2015 Jul;18(4):422–7. PubMed Europe PMC Scholia