Urea cycle and associated pathways (WP4595)
Homo sapiens

{kind=link}
{kind=link}
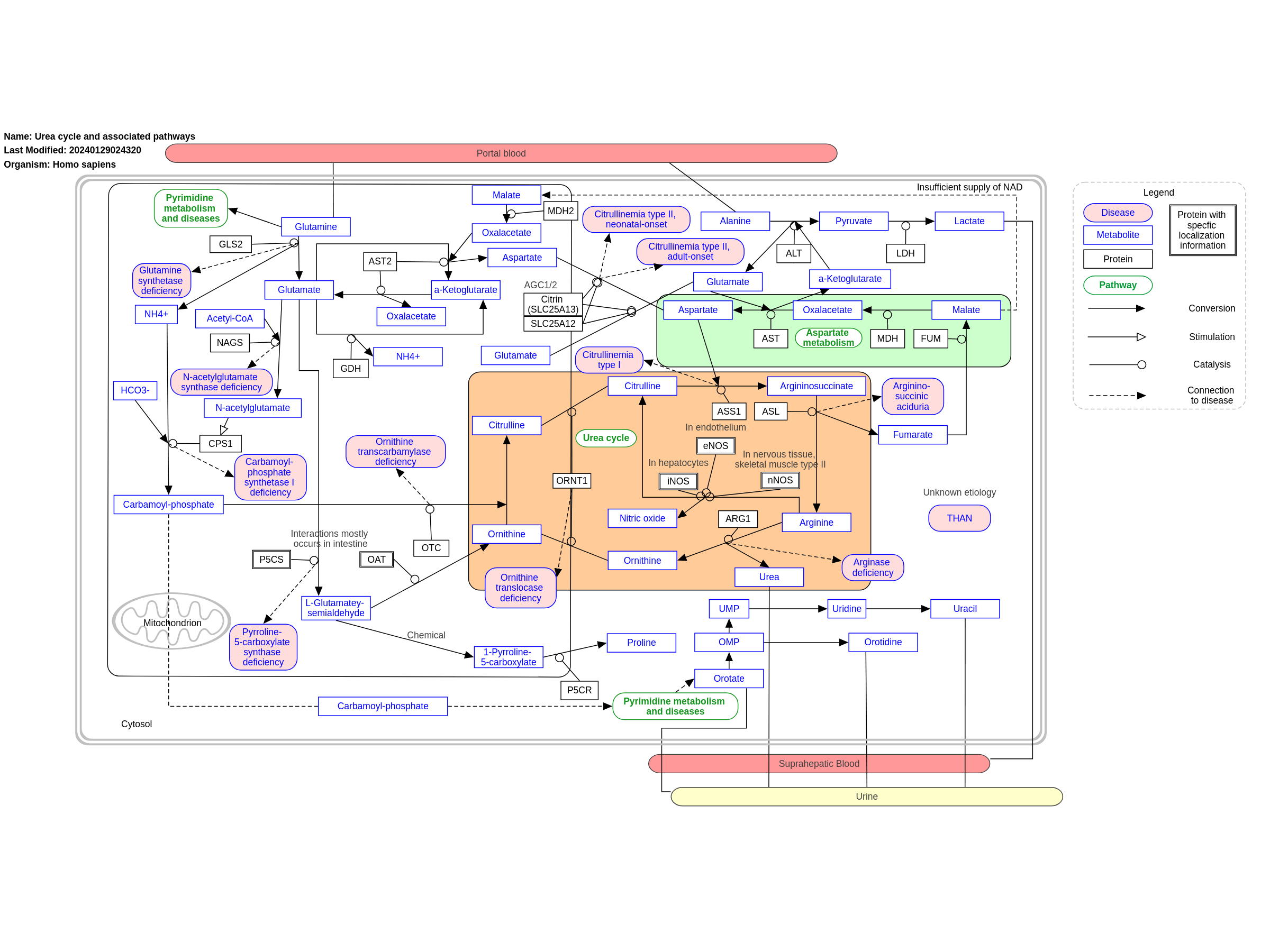
The urea cycle converts toxic nitrogenous compounds to excretable urea in five biochemical reactions. It is also the source for endogenous arginine, ornithine and citrulline production. The process mainly takes place in the liver, partly in the mitochondria and partly in the cytoplasm of the hepatocytes. There are several pathways associated with the urea cycle and with the associated disorders, parts of these pathways are also pictured here. Because there is no alternative way to convert toxic nitrogenous compounds, defects in the enzymes or transporters can lead to several diseases (diseases highlighted in pink). The diseases are characterised by hyperammonemia, respiratory alkalosis and encephalopathy and the severity of the disease depends on the severity of the defect and the place of the defect in the cycle. Severe forms usually have an onset in infancy, while mild forms can also present in adulthood. This pathway was inspired by Chapter 4 of the book of Blau (ISBN 3642403360 (978-3642403361)). For the Urea cycle without additional pathways see: WP4571
For a description of pathway objects, see the WikiPathways Legend.
Authors
Irene Hemel , Denise Slenter , Friederike Ehrhart , Egon Willighagen , Eric Weitz , and Daniela DiglesActivity

Discuss this pathway
Check for ongoing discussions or start your own.
Cited In
- DNA methylation of ARHGAP30 is negatively associated with ARHGAP30 expression in lung adenocarcinoma, which reduces tumor immunity and is detrimental to patient survival (2021).
- Extending inherited metabolic disorder diagnostics with biomarker interaction visualizations (2023).
Are you planning to include this pathway in your next publication? See How to Cite and add a link here to your paper once it's online.
Organisms
Homo sapiensCommunities
Inherited Metabolic Disorders (IMD) Pathways Mitochondrion ONTOX Rare DiseasesAnnotations
Disease Ontology
argininosuccinic aciduria urea cycle disorder citrullinemia hyperargininemia ornithine carbamoyltransferase deficiency carbamoyl phosphate synthetase I deficiency diseasePathway Ontology
inborn error of urea cycle pathway urea cycle pathway disease pathwayCell Type Ontology
hepatocyte| Label | Type | Compact URI | Comment |
|---|---|---|---|
| a-Ketoglutarate | Metabolite | chebi:16810 | aka 2-oxoglutarate |
| Citrulline | Metabolite | chebi:57743 | Zwitterion needed for conversion to take place |
| 1-Pyrroline-5-carboxylate | Metabolite | chebi:17388 | |
| Aspartate | Metabolite | chebi:29991 | (1-) charge needed for conversion to take place |
| Acetyl-CoA | Metabolite | chebi:57288 | (4-) charge needed for conversion to take place |
| L-Glutamatey-semialdehyde | Metabolite | chebi:58066 | |
| Oxalacetate | Metabolite | chebi:16452 | |
| Argininosuccinate | Metabolite | chebi:57472 | (1-) charge needed for conversion to take place |
| Ornithine | Metabolite | chebi:46911 | (1) charge needed for conversion to take place |
| Urea | Metabolite | chebi:16199 | |
| Proline | Metabolite | chebi:60039 | |
| Glutamate | Metabolite | chebi:29985 | (1-) charge needed for conversion to take place |
| Carbamoyl-phosphate | Metabolite | chebi:58228 | (2-) charge needed for conversion to take place |
| Fumarate | Metabolite | chebi:29806 | (2-) charge needed for conversion to take place |
| NH4+ | Metabolite | chebi:28938 | |
| N-acetylglutamate | Metabolite | chebi:44337 | (2-) charge needed for conversion to take place |
| Pyruvate | Metabolite | chebi:15361 | |
| Lactate | Metabolite | chebi:16651 | |
| Glutamine | Metabolite | chebi:58359 | Zwitterion needed for conversion to take place |
| Alanine | Metabolite | chebi:57972 | |
| Malate | Metabolite | chebi:15589 | |
| HCO3- | Metabolite | chebi:17544 | |
| Arginine | Metabolite | chebi:32682 | (1) charge needed for conversion to take place |
| Orotate | Metabolite | chebi:30839 | |
| OMP | Metabolite | chebi:57538 | Orotidylic acid(3-) charge needed for conversion to take place |
| Uridine | Metabolite | chebi:16704 | |
| UMP | Metabolite | chebi:57865 | (2-) charge needed for conversion to take place |
| Uracil | Metabolite | chebi:17568 | |
| Orotidine | Metabolite | chebi:25722 | |
| Nitric oxide | Metabolite | chebi:16480 | (1) charge needed for conversion to take place |
| ARG1 | Protein | uniprot:P05089 | |
| GLS2 | Protein | uniprot:Q9UI32 | |
| P5CS | Protein | uniprot:P30038 | |
| Citrin(SLC25A13) | Protein | uniprot:Q9UJS0 | AKA CTLN2, SLC25A13, AGC2 |
| ASS1 | Protein | uniprot:P00966 | |
| ASL | Protein | uniprot:P04424 | |
| GDH | Protein | uniprot:P00367 | |
| LDH | Protein | uniprot:P07195 | |
| ALT | Protein | uniprot:P24298 | |
| NAGS | Protein | uniprot:Q8N159 | |
| MDH | Protein | uniprot:P40925 | |
| MDH2 | Protein | uniprot:P40926 | |
| OTC | Protein | uniprot:P00480 | |
| OAT | Protein | uniprot:P04181 | |
| SLC25A12 | Protein | uniprot:O75746 | AKA AGC1 |
| AST2 | Protein | uniprot:P00505 | |
| FUM | Protein | uniprot:P07954 | |
| P5CR | Protein | uniprot:P32322 | |
| CPS1 | Protein | uniprot:P31327 | |
| ORNT1 | Protein | uniprot:Q9Y619 | Exchanges ornithine for citruline |
| AST | Protein | uniprot:P17174 | |
| iNOS | Protein | uniprot:P35228 | 'Nitric oxide synthases (EC 1.14.13.39) (NOSs) are a family of enzymes catalyzing the production of nitric oxide (NO) from L-arginine. NO is an important cellular signaling molecule.' [https://en.wikipedia.org/wiki/Nitric_oxide_synthase#iNOS]. iNOS (inducible) is the protein active in hepatocytes, immune and cardiovascular system.aka NOS2 |
| nNOS | Protein | uniprot:P29475 | 'Nitric oxide synthases (EC 1.14.13.39) (NOSs) are a family of enzymes catalyzing the production of nitric oxide (NO) from L-arginine. NO is an important cellular signaling molecule.' [https://en.wikipedia.org/wiki/Nitric_oxide_synthase#iNOS]. nNOS (neuronal) is the protein active in nervous tissue and skeletal muscle type II.AKA NOS1 |
| eNOS | Protein | uniprot:P29474 | 'Nitric oxide synthases (EC 1.14.13.39) (NOSs) are a family of enzymes catalyzing the production of nitric oxide (NO) from L-arginine. NO is an important cellular signaling molecule.' [https://en.wikipedia.org/wiki/Nitric_oxide_synthase#iNOS].eNOS (endotheiliall) is the protein active in endotheliumAKA NOS3, cNOS |
References
- Purified human erythrocyte pyrroline-5-carboxylate reductase. Preferential oxidation of NADPH. Merrill MJ, Yeh GC, Phang JM. J Biol Chem. 1989 Jun 5;264(16):9352–8. PubMed Europe PMC Scholia
- Clinical and functional characterization of a human ORNT1 mutation (T32R) in the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Camacho JA, Mardach R, Rioseco-Camacho N, Ruiz-Pesini E, Derbeneva O, Andrade D, et al. Pediatr Res. 2006 Oct;60(4):423–9. PubMed Europe PMC Scholia
- Arginase Deficiency. Sun A, Crombez EA, Wong D. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 2004. PubMed Europe PMC Scholia
- Citrin Deficiency. Saheki T, Song YZ. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 2005. PubMed Europe PMC Scholia
- Citrullinemia Type I. Quinonez SC, Lee KN. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 2004. PubMed Europe PMC Scholia
- AGC1/2, the mitochondrial aspartate-glutamate carriers. Amoedo ND, Punzi G, Obre E, Lacombe D, De Grassi A, Pierri CL, et al. Biochim Biophys Acta. 2016 Oct;1863(10):2394–412. PubMed Europe PMC Scholia
- Hyperammonemia crisis following parturition in a female patient with ornithine transcarbamylase deficiency. Kido J, Kawasaki T, Mitsubuchi H, Kamohara H, Ohba T, Matsumoto S, et al. World J Hepatol. 2017 Feb 28;9(6):343–8. PubMed Europe PMC Scholia
- Neonatal-onset carbamoyl phosphate synthetase I deficiency: A case report. Yang X, Shi J, Lei H, Xia B, Mu D. Medicine (Baltimore). 2017 Jun;96(26):e7365. PubMed Europe PMC Scholia
- Argininosuccinic aciduria fosters neuronal nitrosative stress reversed by Asl gene transfer. Baruteau J, Perocheau DP, Hanley J, Lorvellec M, Rocha-Ferreira E, Karda R, et al. Nat Commun. 2018 Aug 29;9(1):3505. PubMed Europe PMC Scholia
- N-Acetylglutamate Synthase Deficiency Due to a Recurrent Sequence Variant in the N-acetylglutamate Synthase Enhancer Region. Williams M, Burlina A, Rubert L, Polo G, Ruijter GJG, van den Born M, et al. Sci Rep. 2018 Oct 18;8(1):15436. PubMed Europe PMC Scholia
- Adult-onset type II citrullinemia: Current insights and therapy. Hayasaka K, Numakura C. Appl Clin Genet. 2018 Dec 12;11:163–70. PubMed Europe PMC Scholia